“A dearth or antivirals” Our current repertoire of U.S. Food and Drug Administration (FDA)-approved antivirals is limited to only nine out of the known 214 human-infecting RNA viruses, and almost all these antivirals target viral proteins. Unlike antivirals that target viral proteins, targeting host cell proteins can disrupt the ability of a virus to replicate, effectively shutting down the host cell “virus production facility”. An ideal host-based antiviral would selectively affect only virally infected cells, while leaving uninfected cells unscathed. But how could this work?
The key is that virally infected cells are different than the uninfected cells.
In infected cells, host cell proteins are called to action by the virus to ensure viral replication. In this process, they often leave their normal post, so their normal functions are compromised. We call this a viral-induced hypomorph, which is analogous to a loss-of-function mutation. In cancer, where cells have cancer driver loss-of-function mutations, it is possible to selectively kill cancer cells with drugs that target proteins that are redundant in function to the mutated gene/protein. This is called ‘synthetic lethality’.
Synthetic lethality is a type of genetic interaction between two nonessential genes that participate in a parallel or redundant process to carry out an essential function, where mutations in either gene alone does not affect cell viability, but mutations in both genes result in cell death. We hypothesized that synthetic lethal (SL) interactions of viral-induced hypomorphs could be targeted as host-based antiviral therapeutics – selectively disrupting the viral production facility but leaving uninfected cells unaffected.
Proof of Concept GBF1, a Golgi membrane protein is a critical host factor for many RNA viruses including poliovirus, Coxsackievirus, Dengue, Hepatitis C and E virus, and Ebola virus. GBF1 becomes a hypomorph upon interaction with the poliovirus protein 3A. We performed a genome-wide chemogenomic CRISPR screen using a specific inhibitor of GBF1 called Golgicide A (GCA) in Nalm-6 cells to identify synthetic lethal partners of GBF1 and revealed ARF1 as the top hit. We showed that disruption of ARF1, selectively kills cells that synthesize poliovirus 3A alone or in the context of a poliovirus replicon. Consistent with our hypothesis, combining 3A expression with sub-lethal amounts of GCA further exacerbated the GBF1–ARF1 SL effect.

Extending the principle of synthetic lethal interactions to a virus-induced hypomorph. (A) Synthetic lethality is an extreme negative genetic interaction occurring between two genes. Here, genes ‘A’ and ‘B’ are not essential, and the cell remains viable upon the loss of either gene, depicted by red dotted outline of ‘A’ or ‘B’, individually. However, when these deletions are combined in a single cell, this double loss of function critically impairs the cell, resulting in its death. Such gene-gene combinations are termed synthetic lethal (SL) partners. (B) In the cancerous cell, gene ‘A’ has been mutated, depicted as ‘A*’, leading to an enhanced dependency by the cancer cell for its synthetic lethal partner ‘B’. Drugs that target the otherwise nonessential gene B induce cell death when combined with its SL partner, A*. Therefore, inhibiting the function of B can selectively kill cancerous cells while sparing noncancerous bystander cells. (C) A viral infection provides opportunities for specifically targeting infected cells by synthetic lethality. In an infected cell, host factors, depicted as the red letter ‘A’, are recruited by viral proteins to support viral reproduction. The normal function of the host factor is thus attenuated by the presence of the virus, inducing a hypomorph, red letter ‘A’, which sensitizes the infected cell to inhibition of its synthetic lethal partner by an inhibitory drug.
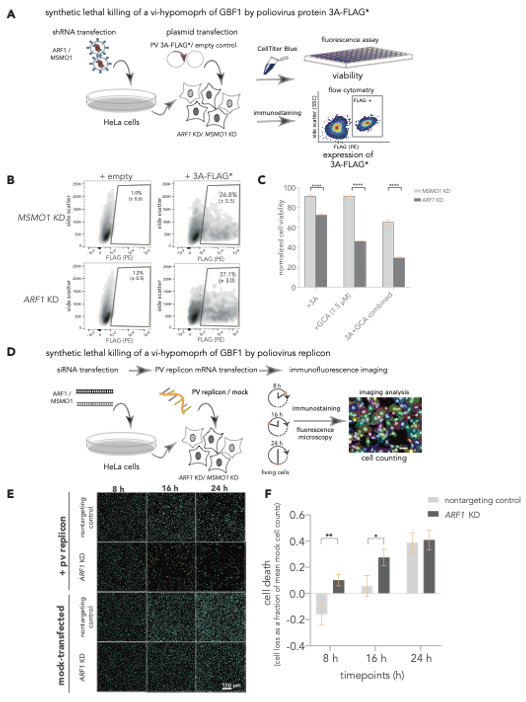
The 3A induced hypomorph of GBF1 sensitizes cells to ARF1-GBF1 synthetic lethality. (A-C) SL killing of a vi-hypomorph of GBF1 by poliovirus protein 3A. (A) ARF1 and MSMO1 were stably silenced in HeLa cells and transfected with FLAG*-tagged poliovirus 3A. Cell viabilities of ARF1 KD and MSMO1 KD cells transfected with 3A-FLAG* or an empty plasmid control were measured with CellTiterBlue. (B) Single cell population (Side scatter (y-axis) positively stained with R-phycoerythrin (PE)-conjugated α-FLAG (x-axis) was quantified to measure the number of cells positive for 3A-FLAG* in ARF1 KD and MSMO1 KD cells by flow cytometry, post 24 h. (C) Poliovirus 3A induces cell death in ARF1 KD cells. Percent viabilities of ARF1 KD and MSMO1 KD cells treated with 3A-alone or GCA-alone (1.5 mM) and a combination of 3A and GCA were measured using CellTiterBlue. A multiple unpaired t-test was used to compare percent viability between the 3A-treated ARF1 KD and MSMO1 KD cells with **** representing P-value < 0.000001. (D-F) SL killing of a vi-hypomorph of GBF1 by poliovirus replicon. (D) HeLa cells were transiently transfected with siRNAs targeting ARF1 and a nontargeting control. The KD or control cells were transfected with 25 ng of poliovirus replicon mRNA, and cells were incubated for 24 h. During the time course of replicon transfection, cells were harvested and fixed at 8, 16, and 24 h, immunostained, and imaged by fluorescence microscopy. (E) Representative immunofluorescence images of nontargeting and ARF1 KD cells transfected with poliovirus replicon or mock. DAPI (cyan) was used to stain cell nuclei and the viral 3A-FLAG* protein was immunostained with a primary antibody against the FLAG tag (orange) (F) Quantification of cell depletion. DAPI signal was used to count the total number of imaged cells. Multiple t-test was used to compare cell death between poliovirus replicon-transfected ARF1 KD and nontargeting controls at each time point. Statistically significant differences were observed at 8 h (P-value < 0.05 indicated as *) and at 16 h (P-value < 0.01, indicated as **).
The Future As viruses co-opt common host factors, SL-derived antivirals have a potential to be broad-spectrum. We established that viral protein interactions can induce hypomorphs that render host cells selectively vulnerable to perturbations that leave uninfected cells otherwise unscathed. Our strategy to target SL interactions of the viral-induced hypomorph has the potential to change the current paradigm for host-based therapeutics that can lead to broad-spectrum antivirals and can be applied to other intracellular pathogens.
This work is supported by National Institutes of Health grants R01 GM112108 and P41 GM109824, R21 AI151344 and foundation grant FDN-167277 from the Canadian Institutes of Health Research.
Access the full text here!